Phlag: Scalable detection of genomics regions with unexplained phylogenetic heterogeneity
Phlag: Scalable detection of genomics regions with unexplained phylogenetic heterogeneity
Sapci, A. O. B.; Arasti, S.; Braun, E.; Mirarab, S.
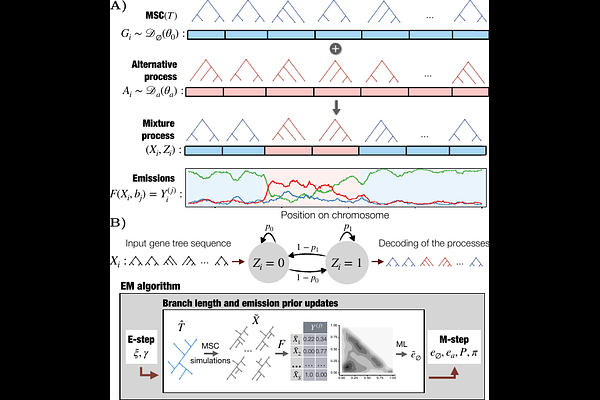
AbstractMotivation: Phylogenetic analyses of entire genomes (phylogenomics) have revealed abundant heterogeneity of evolutionary histories. While much has been done to model this heterogeneity and to infer species trees despite it, the current toolkit has a limitation. Most methods assume that gene trees across the genome differ but are all sampled from the same distribution, defined by models such as the multi-species coalescent (MSC), and parametrized consistently across the genome. Empirical data strongly suggest this assumption is often violated because the species tree, its parameters, or the process generating the gene trees can all change across the genome. Errors in the data can further compound this heterogeneity. To address this challenge, we define the problem of detecting what segments of the genome are inconsistent with a putative species tree, even after allowing discordance according to MSC. We model gene trees not as a set, but rather as a series (a realization of a stochastic process) along genomic positions. We propose a Hidden Markov Model (HMM) approach applied to quartet statistics measured from gene trees and tie the model to MSC using simulations. The combined use of these three ideas leads to a scalable method called Phlag. On simulated and real data, we show that Phlag can detect many cases of change in underlying evolutionary processes, including reduced recombination rates, population size changes, and admixture, all using the same algorithm.