Evolutionary transfer learning enables organism-wide inference of mammalian enhancer landscapes
Evolutionary transfer learning enables organism-wide inference of mammalian enhancer landscapes
Qiu, C.; Daza, R. M.; Welsh, I. C.; Patwardhan, R. P.; Martin, B. K.; Li, T.; Yang, S.; Kempynck, N.; Taylor, M. L.; Fulton, O.; Le, T.-M.; O'Day, D. R.; Lalanne, J.-B.; Domcke, S.; Murray, S. A.; Aerts, S.; Trapnell, C.; Shendure, J.
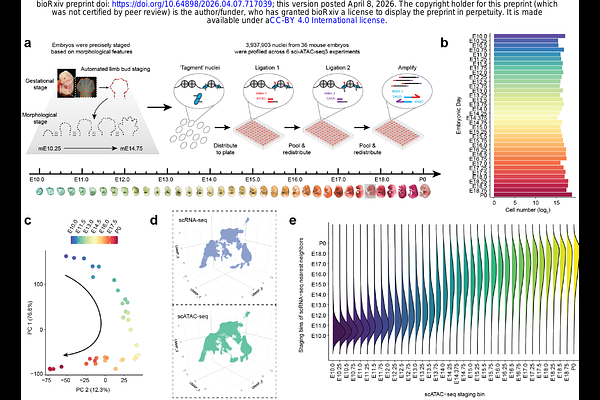
AbstractUnderstanding and modeling how the human genome encodes gene regulatory programs for thousands of cell types remains a central challenge in genomics and machine learning. However, most human cell types emerge during embryonic, fetal, and pediatric development which are inaccessible to comprehensive molecular profiling. To overcome this, we hypothesized that the mismatch in evolutionary rates between cis-acting enhancers (fast) and the trans-acting regulatory programs that interpret them (slow) creates an opportunity for 'evolutionary transfer learning'. Specifically, models trained to predict cell type-specific enhancers in one species should generalize to the orthologous cell types and enhancers of related species. To test this, we generated a single-cell atlas of chromatin accessibility spanning mouse embryonic day 10 (E10) to birth (P0). Using combinatorial indexing[1], we profiled 3.9 million nuclei from 36 staged embryos, resolving genome-wide accessibility in 36 cell classes and 140 cell types. With the goal of identifying distal enhancers for all cell classes, we trained a series of multi-output deep learning models (CREsted[2]), each addressing limitations of the preceding approach. An 'evolution-naive' model achieves strong performance on heldout peaks, but exhibited two failure modes during genome-wide inference: overprediction at tandem repeats and conflation of promoter and distal enhancer grammars. An 'evolution-aware' model resolves these by regrouping accessible regions based on functional coherence across syntenic orthologs, but fails to generalize across species - suggesting insufficient sequence diversity during training. Finally, STEAM (Synteny-aware Transfer learning for Enhancer Activity Modeling), our 'evolution-augmented' model, expands the training corpus to include enhancer orthologs from up to 241 mammalian genomes (Zoonomia[3]) in a synteny-supervised manner. This increases the effective data scale by up to 195-fold, markedly improving generalization across mammals despite greater label noise. We apply STEAM predict enhancers for all major developmental lineages throughout the human, mouse (HumMus) and 239 additional mammalian genomes[3] (BabaGanoush), i.e. 32 x 241 = 7,712 genome-wide enhancer tracks. Together, our results unify advances in single-cell profiling, deep learning, and comparative genomics into a framework for the evolutionary transfer learning of noncoding regulatory grammars. More broadly, our work supports the view that model organisms and evolutionarily diverse genomes are indispensable resources for accelerating the AI-enabled exploration of human biology. Note: An interactive version of this preprint, together with count matrices, CREsted models, prediction tracks, code and reproducible figures, is available at https://doi.org/10.62329/hxkk6249.