Longitudinal study of liver disease progression in the PEX1-Gly844Asp mouse model of mild Zellweger Spectrum Disorder
Longitudinal study of liver disease progression in the PEX1-Gly844Asp mouse model of mild Zellweger Spectrum Disorder
Chen, L.; Choi, H.; Argyriou, C.; Hsieh, M.; Di Pietro, E.; Cui, W.; Nuebel, E.; Daneault, C.; Ruiz, M.; Charpentier, D.; Rhainds, D.; Hacia, J. G.; Nguyen, V.-H.; Gao, Z.; Braverman, N.
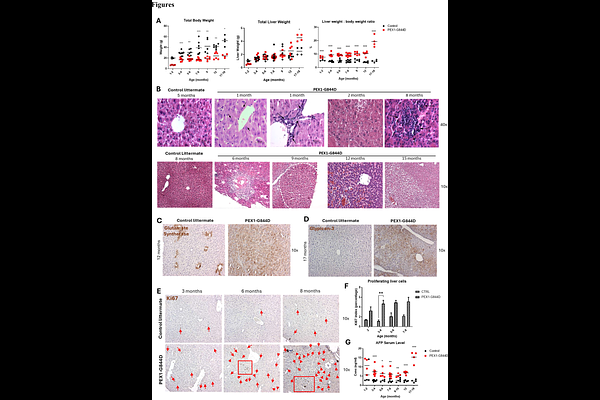
AbstractIntroduction: Zellweger spectrum disorder (ZSD) is an autosomal recessive disorder caused by mutations in any of 13 PEX genes encoding proteins required for peroxisome assembly and function. Chronic liver disease is one of the major clinical manifestations in patients and impacts quality of life and survival. However, the pathophysiology of liver disease is ZSD remains largely unknown, and current interventions are limited. To further study the liver disease mechanism, we use the PEX1-Gly844Asp (G844D) mouse model for mild ZSD, which was previously shown to develop hepatomegaly and cholestasis, similar to ZSD patients. Methods: The natural history of hepatopathy was broadly characterized in PEX1-G844D mice and littermate controls from 1 to 18 months of age using liver histology, electron microscopy, cultured hepatocytes and blood. Metabolite and mechanism analysis included liver functions, respiratory chain dynamics, lipidomics, peroxisome metabolites, gene and protein expression assays. Results: PEX1-G844D mice featured liver disease progression from hepatomegaly (1 month) to cluster cell death (4 months), hepatosteatosis (6 months), inflammation (8 months), fibrosis, and hepatic cancer (12 and 15 months). Hepatocyte proliferation and reduced glycogen was observed across all ages. Measurement of peroxisomal functions showed defective peroxisomal import and secondary mitochondrial defects in cultured hepatocytes. In blood and liver, plasmalogens were decreased, and C26:0 lyso-phosphatidylcholine and C27 bile acid intermediates were elevated. In liver, we observed accumulation of triglycerides and cholesterol, and reduced membrane phospholipids and sphingolipids. In contrast, in serum we observed reduced triglycerides, cholesterol and membrane lipids. Gene expression profiles confirmed by immunoblotting supported reduced hepatic de novo lipogenesis, increased hepatic lipid uptake and oxidation, PPAR activation, and modulated glucose and glycogen metabolism. Liver X receptor agonist (T0901317) applied to cultured hepatocytes enhanced hepatic lipogenesis and lipid secretion, but aggravated steatosis. Conclusion: Taken together, these results suggested the following mechanisms of hepatopathy progression. We propose that global peroxisome dysfunction (1) causes PPAR activation, leading to chronic hyperplasia and partially contributing to disrupted hepatic lipid homeostasis with hepatosteatosis, and (2) underlies chronic hypoglycemia, causing hypoinsulinemia and contributing to reduced hepatic lipogenesis and systemic lipid deficiency. Growth restriction in the mouse model and in ZSD patients could be attributable to systemic lipid deficiency. Our mechanistic delineation of the pathophysiology provides other additional novel potential therapeutic targets to halt liver disease in ZSD.