Disrupted O-GalNAc glycosylation as a mechanism and biomarker of SLC35A2-associated epilepsy
Disrupted O-GalNAc glycosylation as a mechanism and biomarker of SLC35A2-associated epilepsy
Mealer, R. G.; Anderson, J. J.; Smith, S. L.; Masters, B. M.; Barth, S. H.; Huizar, K. D.; Sran, S.; Yoon, H.; Ringland, A.; Muron, S. J.; Bowyer, M. E.; DGama, A. M.; Poduri, A. P.; Lidov, H. G.; Yang, E.; Furnari, J.; Canoll, P. D.; Ostendorf, A. P.; Koboldt, D. C.; Pierson, C. R.; Thomas, D. L.; Philpot, B. D.; Noel, M.; Cummings, R. D.; Heinzen, E. L.; Bedrosian, T. A.
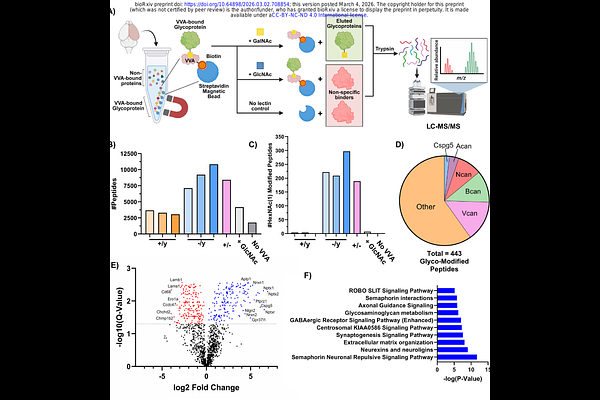
AbstractRare germline and somatic variants in SLC35A2 cause a spectrum of severe glycosylation disorders that commonly present with epilepsy. SLC35A2 encodes the Golgi transporter for UDP-galactose, but how its deficiency leads to severe neurodevelopmental disorders is unknown. Using a mouse model deficient for Slc35a2 in the forebrain, we identified a specific defect in O-GalNAc glycan synthesis, while other galactose-containing glycoconjugates remained intact. O-GalNAc glycans were absent from their normal location within neuronal tracts of the corpus callosum, and truncated precursors accumulated in the cortex on critical extracellular matrix molecules. Cultured primary neurons lacking Slc35a2 showed impaired development, hyperexcitability, and impaired O-GalNAc glycosylation. Finally, human brain tissue from cases of SLC35A2-associated intractable epilepsy displayed a strong correlation between variant burden and truncated O-GalNAc glycans. These findings provide a mechanistic link between genetic causes of SLC35A2-associated epilepsy and protein O-glycosylation that can be targeted for biomarker and therapeutic development.