Col6 deficiency in a zebrafish model of Bethlem myopathy leads to dysfunction of the muscle dihydropyridine receptor
Col6 deficiency in a zebrafish model of Bethlem myopathy leads to dysfunction of the muscle dihydropyridine receptor
Idoux, R.; Exbrayat-Heritier, C.; Shivaraman, S.; Jaque-Fernandez, F.; Ducret, A.; Berthier, C.; Jacquemond, V.; Sohm, F.; bretaud, s.; Ruggiero, F.; ALLARD, B.
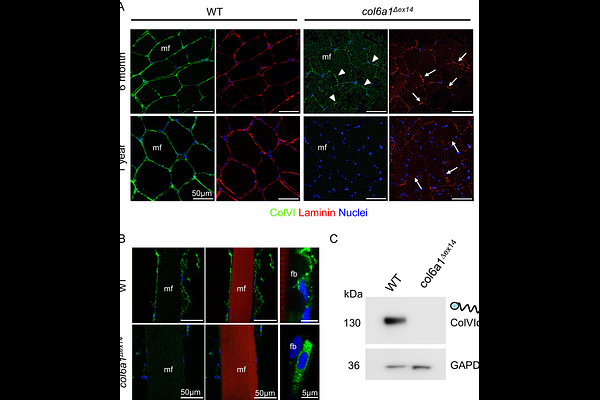
AbstractBethlem myopathy (BM) is an incurable muscle disease characterized by joint contractures and muscle weakness worsening with age. BM results from mutations in genes encoding one of the three chains of collagen VI (ColVI), a component of the skeletal muscle extracellular matrix produced by interstitial fibroblasts. A still unresolved issue in BM is how alteration in ColVI present outside muscle fibers induces dysfunction within muscle fibers. In the present study, we explored properties of excitation-contraction coupling on isolated fast skeletal muscle fibers from one-year old zebrafish col6a1{Delta}ex14 harboring an exon-skipping mutation (col6a1{Delta}ex14) that is the most frequently found in BM patients. Col6a1{Delta}ex14 fish muscle exhibited age-dependent progressive loss of ColVI deposition, accompanied with defects in basement membrane organization and ColVI intracellular accumulation. Muscle action potentials were found to be unchanged in col6a1{Delta}ex14 fish as compared to wild-type. The density of charge movements produced by depolarization-induced activation of dihydropyridine receptors (DHPRs), that control sarcoplasmic reticulum (SR) Ca2+ release, was found to be reduced and their voltage-dependence shifted toward negative potentials in col6a1{Delta}ex14 fish. Concomitantly, the voltage dependence of depolarization-evoked intracellular Ca2+ transients was also shifted toward negative voltages, promoting in this way an elevated pathogenic SR Ca2+ leak at resting membrane potentials, as evidenced by higher density of Ca2+ sparks at rest, larger SR Ca2+ release in response to long-duration depolarizations between -70 and -40 mV and reduced twitch muscle force and swimming performance. Finally, clusters of 1S subunits of DHPR were found to be mis-localized in mutant fibers t-tubules. These data suggest that Col6 deficiency in BM leads to an alteration of the DHPR function that contributes to promote a pathogenic SR Ca2+ leak. DHPR could represent the still elusive link that allows altered myomatrix to transduce pathogenic signals within muscle.