Interleukin-6 is critical in the development of gcn2-mutation associated pulmonary vascular disease in mice
Interleukin-6 is critical in the development of gcn2-mutation associated pulmonary vascular disease in mice
Schwiening, M.; Gao, Q.; Southwood, M.; Crosby, A.; Moore, S.; Valer, J. A.; Veale, N.; Dunmore, B.; Upton, P. D.; Thompson, R.; Morrell, N.; Marciniak, S. J.; Soon, E.
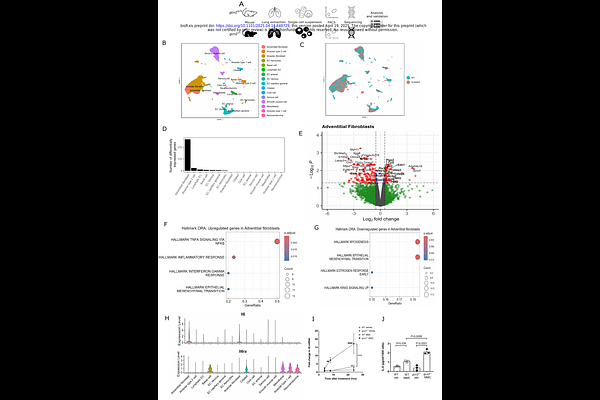
AbstractBiallelic mutations in eukaryotic translation initiation factor 2 kinase 4, EIF2AK4 (which encodes general control nonderepressible 2, GCN2) underpin heritable forms of pulmonary veno-occlusive disease (PVOD), a rare and fatal form of pulmonary hypertension. The mechanisms linking these are mostly uncharacterised. We demonstrate for the first time that homozygous loss of gcn2 is sufficient to cause mild pulmonary hypertension in mice. Single-cell transcriptomics of mouse lungs identified adventitial fibroblasts as having the greatest GCN2-dependent transcriptional differences, implicating them as key players in this model of PVOD. The most significantly upregulated pathways in gcn2-/- adventitial fibroblasts were inflammatory. Therefore, we went on to demonstrate a pro-inflammatory phenotype in gcn2-/- mouse embryonic fibroblasts and gcn2-/- mice. In a novel murine model of pulmonary hypertension induced by exposure to mitomycin C, deletion of interleukin-6 rescued the pulmonary vascular phenotype. When chronically exposed to lipopolysaccharide, the pulmonary hypertensive phenotype of gcn2-/- mice is exaggerated. Genetic ablation of interleukin-6 completely rescues both the baseline and LPS-exaggerated pulmonary hypertensive phenotype. Targeting Il6-dependent pathways may be useful in treating this deadly disease.