Therapy resistance in AML is mediated by cytoplasmic sequestration of the transcriptional repressor IRF2BP2
Therapy resistance in AML is mediated by cytoplasmic sequestration of the transcriptional repressor IRF2BP2
Althoff, M. J.; Minhajuddin, M.; Stevens, B.; Gillen, A. E.; Gipson, S.; Patel, S. B.; Shelton, I. T.; Miller, R.; Vujovic, A.; Krug, A.; Young, T.; Showers, W. M.; Dzieciatkowska, M.; Stephenson, D.; Tyagi, A.; Ellegast, J. M.; Wright, T.; Stegmaier, K.; Opferman, J. T.; D'Alessandro, A.; Smith, C. A.; Jordan, C. T.
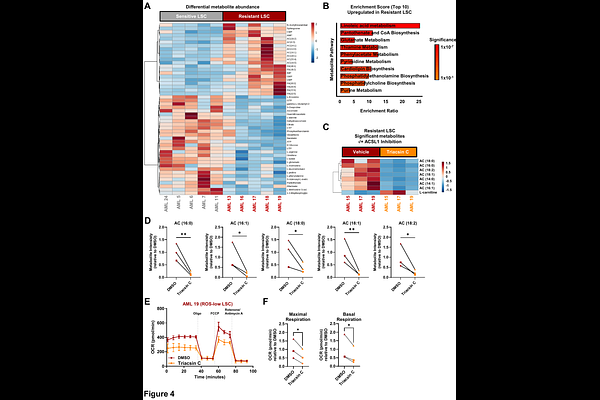
AbstractWhile the development of venetoclax with azacitidine (ven/aza) has improved AML therapy, drug resistance remains a major challenge. Notably, primary ven/aza-resistant AML are frequently reliant on MCL1, however, the underlying mechanisms remain unclear. Co-immunoprecipitation of MCL1 from ven/aza-resistant AML samples coupled with mass spectrometry analysis identified the transcriptional repressor Interferon Regulatory Factor 2 Binding Protein 2 (IRF2BP2) as an MCL1 binding partner. This interaction results in cytoplasmic IRF2BP2 localization and loss of transcriptional repression within ven/aza-resistant leukemic stem cells (LSC). Consequently, ven/aza-resistant LSC have increased IRF2BP2 target gene expression, including acyl-CoA synthetase long-chain family member 1 (ACSL1), an essential rate-limiting enzyme for fatty acid oxidation (FAO). Inhibition of ACSL1 functionally impaired ven/aza-resistant LSC through a depletion of long-chain acyl-carnitine metabolites and FAO. Collectively, these data provide evidence for a previously undescribed mechanism by which MCL1 mediates IRF2BP2 cytoplasmic sequestration and consequent de-repression of ACSL1, thereby promoting ven/aza-resistance in AML.