In vivo anti-tumor activity of high-dose parenteral ascorbic acid is mediated primarily via cofactor activity, not via oxidative stress.
In vivo anti-tumor activity of high-dose parenteral ascorbic acid is mediated primarily via cofactor activity, not via oxidative stress.
Akram, T.; Luchtel, R. A.; Dubey, V.; Seal, S.; Aggarwal, R.; Sai, H.; Shenoy, N. K.
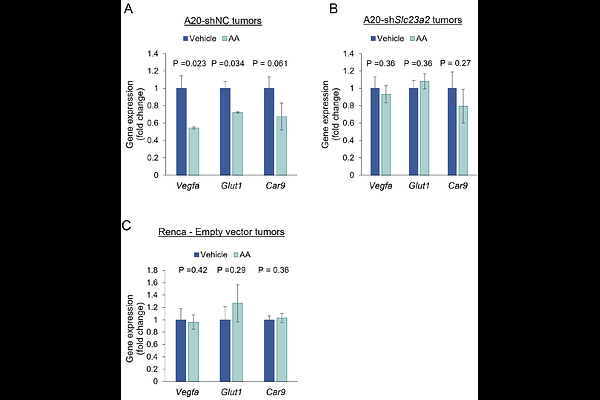
AbstractThe anti-tumor effect of high-dose ascorbic acid (AA) has been demonstrated in multiple in vitro and in vivo cancer models with the postulation of two primary categories of mechanisms: antioxidant/cofactor activity and H2O2-mediated oxidative damage. Both mechanisms have been conclusively demonstrated in vitro. However, while parenteral high-dose AA-induced cofactor activity (TET-mediated DNA demethylation and prolyl/ asparaginyl hydroxylase-mediated HIF activity inhibition via reduction of enzymatic Fe3+ to Fe2+) has been demonstrated intratumorally in vivo in multiple models, the cumulative data on parenteral high-dose AA-induced intratumoral oxidative damage in vivo has been inconclusive. Furthermore, the relative contribution of the seemingly opposing mechanisms towards in vivo anti-cancer activity has not been studied concurrently. We therefore sought to definitively delineate the roles of both antioxidant/ cofactor activity and prooxidant functions of high-dose AA in the in vivo anti-tumor response. Using two syngeneic mouse tumor models, the AA-sensitive A20 model and the AA-resistant Renca model, we assessed markers of DNA and lipid oxidative damage as well as the specific roles of TET2 and AA transporter SLC23A2 in the anti-tumor response to parenteral high-dose AA. In the sensitive A20 model, loss of either Tet2 or Slc23a2 fully reversed anti-tumor activity. Similarly, overexpression of Tet2 in the resistant Renca model (which expresses high baseline levels of AA transporters SLC23A1 and SLC23A2, but does not express TET2), resulted in increased CD8+T cell infiltration and dramatic reduction in tumor growth overall. In both A20 and Renca models, high-dose parenteral AA increased total intratumoral antioxidant capacity, and this was attenuated by Slc23a2 knockdown in A20. High-dose AA treatment also resulted in a Tet2- and Slc23a2-dependent increase in intratumoral 5-hydroxymethylcytosine. Intracellular oxidative damage markers, 8-OHdG and 4-HNE, were not induced in tumors by high-dose AA in either model. In contrast, these markers were robustly induced in vitro by high-dose AA in A20 and Renca cells. Using dynamic real-time extracellular H2O2 measurements with high-dose AA, difference in molecular oxygen concentration between standard in vitro and hypoxic in vivo conditions was identified as an important factor underlying the marked discrepancy between the abundant in vitro and absent in vivo intratumoral oxidative stress with high-dose AA. Furthermore, using additional syngeneic models resistant (MB49) and sensitive (MC38) to AA-induced potentiation of anti-PD1 checkpoint inhibition, we demonstrate that very low catalase expression does not confer sensitivity to high-dose AA in vivo (further arguing against the H2O2 mechanism in vivo), that TET2 expression alone is not sufficient to drive an AA-induced anti-tumor response (either as a single agent or in combination with immunotherapy), and that high-dose AA can significantly enhance the efficacy of anti-PD1 immunotherapy even in the absence of single-agent activity. Our data strongly indicate that the in vivo anti-tumor effect of high-dose parenteral AA- including potentiation of immunotherapy- is mediated primarily by its specific antioxidant/ cofactor activity (with TET2 expression likely being necessary but certainly not sufficient), and not via oxidative stress. Collectively, the study represents a paradigm shift in our understanding of the cumulative mechanisms of in vivo anti-cancer activity of high-dose AA, with critical implications not just for the clinical translation of AA as an anti-cancer agent (including in enhancing immunotherapy efficacy) but also the field of free radical biology.