Benchmarking Docking Tools on Experimental and Artificial Intelligence-Predicted Protein Structures
Benchmarking Docking Tools on Experimental and Artificial Intelligence-Predicted Protein Structures
Tejera Nevado, P.; Junod, N.; Hyunjin Kwon, E.; Prieto Santamaria, L.; Rodriguez Gonzalez, A.
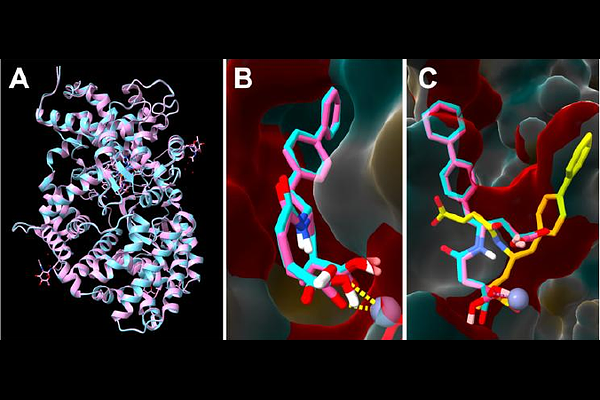
AbstractIn silico analysis provides valuable insights into studying macromolecules, particularly proteins. Protein structure prediction models, like AlphaFold (AF), offer a cost-effective and time-efficient alternative to traditional methods like X-ray crystallography, NMR spectroscopy, and cryo-EM for determining protein structures. These models are increasingly used in protein-ligand interaction studies, a key aspect of drug discovery. Docking and molecular dynamics simulations facilitate this process, and researchers are continuously developing open-access tools for cavity detection and docking to accelerate protein-ligand interaction studies. However, while many of these tools perform well in specific cases, their strengths and weaknesses in analyzing predicted protein structures remain largely unknown. Therefore, it is crucial to compare docking analyses using experimentally determined protein structures and deep learning-based models. In this study, two well-characterized proteins, dopamine D3 receptor with its ligand ETQ and neprilysin with its ligand sacubitrilat, are used to evaluate docking predictions. The docking tools CB-Dock 2 and COACH-D are applied to both X-ray crystallography-derived structures and five different AFgenerated models. The objective is to assess the accuracy of these docking approaches and determine whether this strategy can effectively simulate macromolecular behavior in their microenvironment. By doing so, this study aims to generate new insights and contribute to accelerating research in protein-ligand interactions.