A Machine Learning Approach for Physiological Role Prediction in Protein Contact Networks: a large-scale analysis on the human proteome
A Machine Learning Approach for Physiological Role Prediction in Protein Contact Networks: a large-scale analysis on the human proteome
Cervellini, M.; Martino, A.
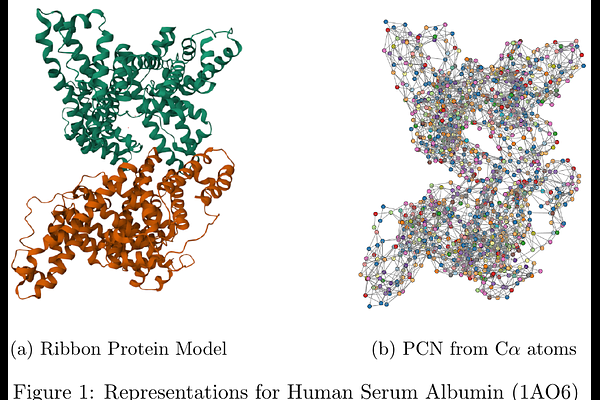
AbstractProteins are fundamental macromolecules involved in virtually all biological processes. Their physiological roles are tightly linked to their three-dimensional structure, which can be naturally abstracted as Protein Contact Networks (PCNs), i.e., graphs where residues are nodes and edges encode spatial proximity. This representation enables the application of Graph Machine Learning to address the protein functional annotation gap at proteome scale. In this work, protein function prediction is studied on the majority of the human proteome, focusing on enzymatic activity and enzyme class assignment as well-defined and biologically meaningful targets. A large-scale supervised analysis was conducted on PCNs derived from experimentally resolved human protein structures. Multiple graph-based learning paradigms were systematically compared under a unified evaluation protocol, including handcrafted graph embeddings, kernel methods, and end-to-end Graph Neural Networks (GNNs). Feature engineering approaches comprised (i) spectral density embeddings of the normalized graph Laplacian and (ii) higher-order topological representations based on simplicial complexes, with optional INDVAL-based feature selection. These representations were paired with linear, ensemble, and kernel classifiers, while GNNs were trained directly on raw PCNs exploiting a diverse set of message-passing architectures. Two tasks were considered: binary classification of enzymatic versus non-enzymatic proteins and multiclass prediction of first-level Enzyme Commission (EC) classes. Performance was assessed using repeated stratified splits to ensure robust and variance-aware evaluation. In the binary enzymatic classification task, the Jaccard-based graph kernel achieved the best performance with an adjusted balanced accuracy of 0.90, closely followed by GNNs trained end-to-end on PCNs. In the multiclass EC prediction task, GNNs demonstrated superior discriminative power, reaching an adjusted balanced accuracy of 0.92 and outperforming all explicit embedding and kernel-based approaches. Overall, results indicate that EC class prediction is intrinsically more complex than binary enzymatic discrimination and benefits from the higher expressivity of deep message-passing architectures. The findings demonstrate that graph-based representations of protein structure support competitive functional prediction at proteome scale, with classical kernel methods and modern GNNs offering complementary strengths in terms of accuracy, scalability, and flexibility.