Dcaf17 knockdown integrates mitochondrial-lysosomal dysfunction with ferroptosis, NLRP3 inflammasome signaling and necroptosis
Dcaf17 knockdown integrates mitochondrial-lysosomal dysfunction with ferroptosis, NLRP3 inflammasome signaling and necroptosis
Alyacoub, n.; Almohanna, F.; Alqassem, A.; Awad Mahmoud, S.; Assiri, A. m.
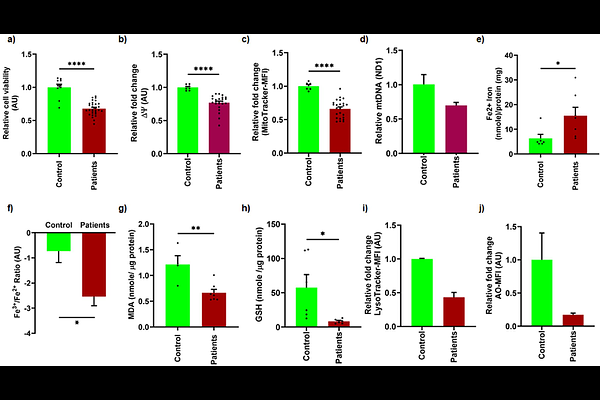
AbstractDCAF17 (DDB1- and CUL4-associated factor 17) plays a vital role in spermatogenesis, influencing post-meiotic sperm development. Its dysfunction leads to male infertility, as demonstrated in our previous study on Dcaf17 knockout mice, which revealed impaired sperm morphogenesis, increased numbers of dysmorphic and immotile sperm, and complete male infertility. Mutations in the human Dcaf17 gene have been implicated as causative factors for Woodhouse-Sakati Syndrome (WSS), a disorder characterized by hypogonadism and infertility, further emphasizing its critical role in male reproductive function. Despite these observations, the molecular mechanisms underlying DCAF17 function remain poorly defined. Here, we employed the GC1 spermatogonia cell line to investigate the role of DCAF17 in germ cell viability and homeostasis. Lentiviral shRNA-mediated knockdown of Dcaf17 significantly decreased cell viability and increased cell death. Functional studies demonstrated a notable decline in mitochondrial membrane potential and mass, coinciding with increased oxidative phosphorylation (OXPHOS) protein expression. Unexpectedly, both mitochondrial and cytosolic reactive oxygen species (ROS) levels were reduced following Dcaf17 silencing. Further investigation uncovered profound lysosomal damage, evidenced by accumulation of SQSTM1/p62 and loss of acidity. Transcriptional and translational profiling indicated downregulation of key iron transport and antioxidant genes (xCT, transferrin), while levels of transferrin receptor were elevated. Silencing of Dcaf17 triggered ferroptosis, as evidenced by elevated Fe2+, reduced glutathione (GSH) and increased lipid peroxidation (MDA). In parallel, inflammatory and necroptotic pathways were activated, as indicated by upregulation of MLKL, NLRP3, cleaved IL-1 {beta}, and phosphorylated JNK1/2, along with increased LDH release and reduced Bcl-xL and Bcl-2. These molecular signatures were recapitulated in lymphoblastoid cell lines from WSS patients with a pathogenic Dcaf17 mutation, which exhibited mitochondrial and lysosomal dysfunction, disrupted iron homeostasis, and oxidative stress. Similar alterations were observed in testis tissue and sperm from Dcaf17 knockout mice, including increased Fe2+ and MDA levels, reduced GSH, and impaired mitochondrial function in sperm. Collectively, our findings establish DCAF17 as a key regulator of mitochondrial integrity, lysosomal function, iron metabolism, and regulated cell death pathways in spermatogonia. This study provides the first mechanistic insight into how Dcaf17 deficiency contributes to male infertility.