LLMsFold: Integrating Large Language Models and Biophysical Simulations for De Novo Drug Design
LLMsFold: Integrating Large Language Models and Biophysical Simulations for De Novo Drug Design
Waththe Liyanage, W. W.; Bove, F.; Righelli, D.; Romano, S.; Visone, R.; Iorio, M. V.; Lio, P.; Taccioli, C.
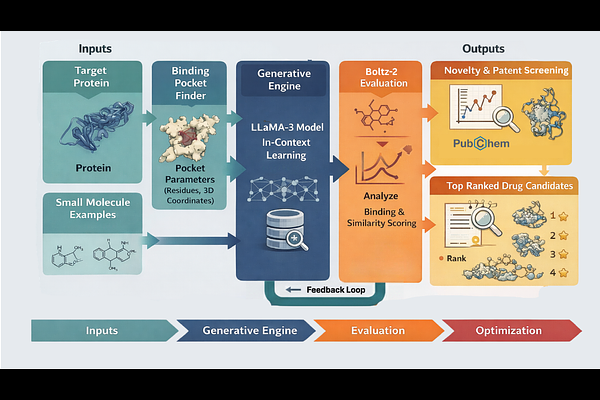
AbstractThe discovery of novel small molecules is challenging because of the vastness of chemical space and the complexity of protein-ligand interactions, leading to low success rates and time-consuming workflows. Here, we present LLMsFold, a computational framework that combines Large Language Models (LLMs) and biophysical foundation tools to design and validate new small molecules targeting pathogenic proteins. The pipeline starts by identifying viable binding pockets on a target protein through geometry-based pocket detection. A 70-billion-parameter transformer model from the LlaMA family then generates candidate molecules as SMILES strings under prompt constraints that enforce drug-likeness. Each molecule is evaluated by Boltz-2, a diffusion-based model for protein-ligand co-folding that predicts bound 3D structure and binding affinity. Promising candidates are iteratively optimized through a reinforcement learning loop that prioritizes high predicted affinity and synthetic accessibility. We demonstrate the approach on two challenging targets: ACVR1 (Activin A Receptor Type 1), implicated in fibrodysplasia ossificans progressiva (FOP), and CD19, a surface antigen expressed on most B-cell lymphoma and leukemia cells. Top candidates show strong in silico binding predictions and favorable drug-like profiles. All code and models are made available to support reproducibility and further development.